


The most common hereditary disease in the white race!
Cystic Fibrosis (CF) is an incurable, particularly life threatening, genetic, hereditary, non-transmittable disease that causes death at an early age.
This fact makes Cystic Fibrosis the major hereditary disease in our country as, due to the population screening for thalassaemia, eventhough the carrier rate is estimated at around 8%, the births of children with thalassaemia have been significantly reduced.
The life expectancy of individuals with cystic fibrosis is from 2 to 47 years with the survival rate being increased in advanced societies with good health care, but it does not cease the fact of moderate to poor quality of life.
For more information on Cystic Fibrosis see the brochure here…

Cystic Fibrosis is a hereditary disease due to a CFTR (Cystic Fibrosis Transmembrane Regulator) gene mutation in the 7th chromosome. Over 1,900 different mutations and polymorphisms have been observed leading to the same disease, but the most common is ΔΦ508. The overwhelming majority of mutations have a very low occurrence rate below 0.1% of the population, with a large heterogeneity in the distribution of mutations among different nationalities.
For a person to have a Cystic Fibrosis, both parents must be carriers and the person inherit two defective genes – one from each parent. Thus, whenever a child is captured by two cystic fibrosis vectors, there are the following possibilities:
- 25% chance that their child will not be a patient or even a carrier.
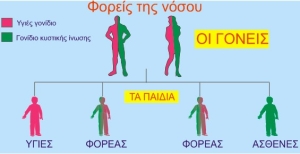
- 50% chance that the child will be a carrier of the gene without any other effect and
- 25% chance that the child will have Cystic Fibrosis

These percentages apply to any pregnancy regardless of the sex of the child.
If one parent is affected and the other one is a carrier then in every pregnancy there is a 50% chance that the child is a carrier and 50% likelyhood to be affected.
If one parent is affected and the other is not a carrier, then all children will be carriers.
If one parent is the carrier and the other negative for 90% of the mutations, the probability of having a child is 1/960 (0.10%).
In Greece, population screening of cystic fibrosis is not mandatory, but it is better if it is possible to screen at least one parent and definitely to screen couples having cystic fibrosis because there is no cure for this disease.
The most common, simple and fast way to diagnose patients is the so-called sweat test with the Gibson-Cooke method, which measures the concentration of sodium and/or chlorine in the sweat, the collection of which is a very simple procedure. Values from 0 to 40 mEq/L are considered normal, from 40 to 60 mEq/L in the boundary area, while for values above 60 mEq / L the test is considered positive. Two continuous tests are usually performed to confirm the diagnosis.
It should be noted that there are rare mutations that give values in the negative or marginal area, but nevertheless the patient suffers from Cystic Fibrosis.
Due to the large number of mutations leading to the same disease and the heterogeneity that occurw according to nationality, the genetic analysis of all mutations is relatively expensive and therefore the most frequent choice is to study the most common mutations observed in each population.
At Safembryo 3 mutation analysis options are available for the KIN:
- ΔF508 (F508del): Includes analysis of the most common mutation which is however found only in 53% of all patients in Greece
- CF 85%: Includes analysis of the most frequent mutations that cover 85% of known mutations in the Greek population.
The reagent system used covers most of the known cystic fibrosis mutations in Greece and is officially licensed for diagnostic purposes (CE-IVD).

- CF> 98%: Includes control of the entire CFTR gene including duplications and deletions which detects> 99% of the mutations in the gene.
This is a simple peripheral blood test where the results are available within one to three weeks of extensive screening.
- Couples who plan to make or wait for a child
- Family members with relatives with CF
- Partners of patients or patients with CF
- Fetal ultrasound findings (eg echogenic bowel) indicating increased risk for CF
- Men with sterility due to lack of sperm, obstructive azoospermia and oligospermia (In these individuals the presence of cystic fibrosis in heterozygous at least form is calculated at a rate ranging from 30-70%).
- In people with unspecified Chronic Pulmonary Disease.
A positive result means there is a copy of a mutation that is known to cause CF in your DNA. This does not mean you have CF. If it is found that you are a CF carrier, then your partner will have to be checked. If it turns out that both of you are CF carriers, your doctor or genetic counselor will discuss with you the options you have for reproduction and prenatal screen.
A negative result greatly reduces the risk of being a CF carrier (depending on the test option selected), but it does not diminish it. Since this test does not detect all mutations that cause CF, and because all CF mutations may not yet be known, a negative result can not completely eliminate the likelihood of being a carrier.
Typical symptoms of CF

- Persistent and unjustified cough.
- Common respiratory infections.
- Inability to gain weight.
- Repeated diarrhea.
- Extremely salty sweat.
Symptoms vary from person to person, partly due to more than 1,900 mutations of the CF gene.
Cystic fibrosis affects mainly the lungs, the pancreas, the liver and the intestine. It is characterized by the abnormal transfer of chloride and sodium ions through the epithelium resulting in the production of particularly dense mucus which blocks the various organs and pores of the body, mainly the lungs and the pancreas, resulting in severe pancreatic insufficiency from a very young age and appearance severe chronic respiratory infections that gradually destroy the lungs and lead the patient to respiratory failure and death.
The disease affects many other organs in the body, such as:
- the liver with cirrhosis,
- sinuses with the appearance of polyps and sinusitis from a very young age,
- bile secretion in the liver, eventually causing permanent liver damage in about 6% of Cystic Fibrosis patients.
- bones and joints with the development of rheumatoid arthritis, osteopenia and osteoporosis,
- the genital system in men who, overwhelmingly, face fertility problems,
- the intestines with the creation of ileus and sweat glands.
- Due to pancreatic insufficiency, patients are difficult to put on weight and often have diabetes.
Treatment of CF depends on the stage of the disease and the organs involved. The primary goal treatment is to treat respiratory infections that lead to respiratory failure.
From the time of diagnosis, a Cystic Fibrosis patient should be monitored and hospitalized in a specialized Cystic Fibrosis Center. For the elimination of bronchial secretions and the release of airways from the dense mucus, it is subjected daily to long-term and tiring respiratory physiotherapy.
Chest physiotherapy is a form of cleansing of the respiratory corridor that is done by a powerful blow over the back and chest in order to extract the thick mucus from the lungs.
In addition,  about 90% of people with CF before each meal receive a large number of enzymes, vitamins, insulin and other pills to help them absorb food nutrients because they suffer from severe pancreatic insufficiency.
about 90% of people with CF before each meal receive a large number of enzymes, vitamins, insulin and other pills to help them absorb food nutrients because they suffer from severe pancreatic insufficiency.
It is absolutely necessary to take daily inhalation therapy and mucolytics and bronchodilators that help it to stop pulmonary infections and improve lung function.
Periodically, sufferers are subjected to extremely potent intravenous antibiotic therapies, remaining for some days in the hospital to cope with chronic pulmonary infections that disturb them as they are prone to them.
So far, there is no definitive treatment for this multi-systemic disease and, in some cases, the patient is in the final stages of respiratory failure, is undergoing lung transplantation if a histocompatible donor is found.